
Nat Metab:厦门大学林圣彩团队揭示葡萄糖感知通路的关闭是维持大脑髓鞘修复的关键
时间:2025-10-26
来源:iNature 2025-10-26 17:04
少突胶质前体细胞拥有一套独特的分子“刹车”系统,能够在脱髓鞘所引起的病灶区设法不抑制mTORC1来维持合成代谢,以保障髓鞘的重新形成。2025年10月22日,河南省中州实验室、河南大学基础医学院,连同厦门大学林圣彩院士团队在Nature Metabolism上发表题为Oligodendrocyte precursor cell-specific blocking of low-glucose-induced activation of AMPK ensures myelination and remyelination的文章,揭示了负责形成神经保护层 髓鞘的 绝缘材料 细胞,即少突胶质前体细胞(oligodendrocyte precursor cells, OPCs),拥有一套独特的分子 刹车 系统,能够在脱髓鞘所引起的病灶区 此处的葡萄糖水平显著降低,设法不抑制mTORC1来维持合成代谢,以保障髓鞘的重新形成。这一发现为理解多发性硬化症、阿尔茨海默病等与脱髓鞘密切相关的神经系统疾病的发病机制,以及开发潜在治疗策略提供了全新视角。

低糖危机下的负有特殊 使命 细胞,为修复备战而 无视 警报
与肝脏、肌肉等外周组织类似,葡萄糖也是大脑的主要碳源。先前的研究表明,在外周组织中,葡萄糖水平降低会通过细胞内的溶酶体途径,激活AMPK(AMP-activated protein kinase, AMPK),抑制消耗ATP的合成代谢,并启动分解代谢,以一种 节能保供 的模式保证组织不会遭受 能源危机 。然而,作者们惊讶地发现,大脑中的OPC显得 特立独行 即使在低糖环境中,其AMPK也保持 沉默 ,不会被激活,这和其他种类的脑细胞大不相同。他们进一步发现,OPC的这种 钝感 具有重要的生理意义:OPC核心使命是增殖并分化为成熟的少突胶质细胞,后者通过形成包裹神经纤维的髓鞘(myelination),确保神经信号高速、精准地传递。这一过程主要发生在大脑的发育阶段,以及成体髓鞘损伤后的修复期(又被特指为 髓鞘再生 ,即remyelination),但无论是哪个时期,OPC都需合成大量蛋白质和脂质。如果OPC像其他细胞一样在低糖时激活AMPK,其合成代谢将被抑制,增殖与分化过程便会受阻,导致髓鞘形成或再生的障碍,进而引发严重的神经系统疾病。
分子机制揭秘:一个乙酰化修饰如何决定细胞命运
作者们进一步探究为何OPC中的AMPK不在低糖条件下被激活。他们发现,OPC中的溶酶体AMPK途径被 钝化 了。具体地说,OPC中介导溶酶体途径激活的关键分子TRPV(transient receptor potential V)在低糖条件下依然保持活力,其下游分子、位于溶酶体上的质子泵v-ATPase也保持活力。本应在此时以v-ATPase及Ragulator作为锚定点被募集到溶酶体表面的的架构蛋白AXIN也不再定位于溶酶体,而是继续留在细胞质中;这导致AMPK的上游激酶、也是依赖于AXIN才能被转运到溶酶体附近的LKB1(kinase liver kinase B1)无法接近定位在溶酶体的AMPK,从而无法将低糖的信号转化成AMPK的激活。
作者们于是进一步探究溶酶体途径是如何被 钝化 的。他们之前已发现,低糖是经由溶酶体上的醛缩酶(aldolases;由ALDOA-ALDOC三种isozymes组成)所感知。更准确地说,处于无FBP结合的 空闲(idle) 状态的醛缩酶能感知了葡萄糖的糖酵解中间产物果糖-1,6-二磷酸(fructose-1,6-bisphosphate, FBP)的水平下降,并抑制了TRPV,进而激活了溶酶体途径和AMPK。这说明,在OPC中,AMPK激活的阻断很有可能是FBP的感知器醛缩酶发生了变化。进一步的研究发现,这其中的关键在于OPC中表达量最高的ALDOC的第14位赖氨酸发生了乙酰化修饰。这个修饰如同一个 绝缘开关 ,使得醛缩酶在低糖/FBP时无法结合和抑制TRPV。进一步验证发现,该位点突变为模仿 去乙酰化 状态的醛缩酶(ALDOC-K14R,即ALDOC第14位的赖氨酸突变为精氨酸),能恢复OPC感知低糖并激活AMPK的能力。例如成熟后的少突胶质细胞(mature oligodendrocytes,mOL);OPC一旦分化成成熟的少突胶质细胞(mature oligodendrocytes,mOL)后,去乙酰化酶SIRT2表达水平显著升高,将醛缩酶上的乙酰基团去除,便移除了这个 绝缘 机制,因此mOL可以正常响应葡萄糖波动(图1)。
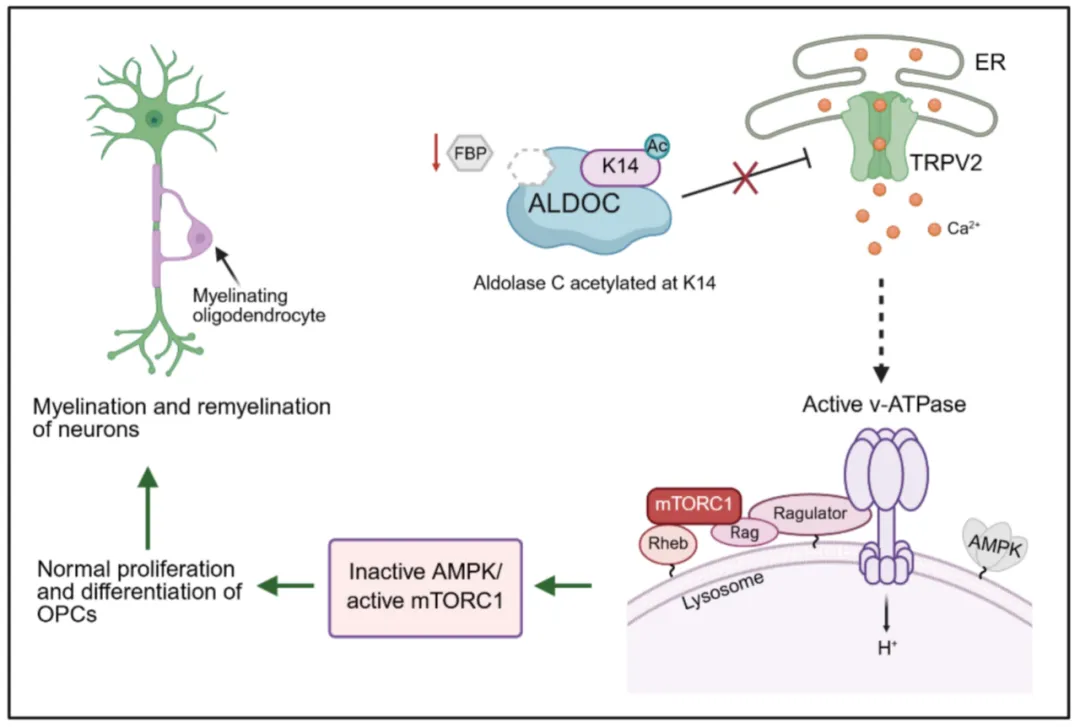
图1 OPC中的ALDOC-K14的乙酰化 钝化 了溶酶体途径感知低糖、激活AMPK并抑制mTORC1的能力,从而保证了OPC在髓鞘修复时所需的高强度的合成代谢
从机制到疾病模型:干预 开关 影响神经元修复能力
为了验证这一钝化AMPK激活的生理意义,作者们构建了多种脱髓鞘疾病模型(如Cuprizone、LPC模型及EAE模型)。他们发现,在这些模型中,脱髓鞘导致了该区域内葡萄糖浓度的显著下降,而且也确认了AMPK不被激活。而在OPC特异性表达ALDOC-K14R的小鼠中,AMPK在脱髓鞘区域的OPC中被激活,其增殖和分化严重受损,导致髓鞘再生障碍,疾病症状显著加重(图1)。
作者还进一步验证了不被乙酰化的醛缩酶突变体ALDOC-K14R在OPC的表达导致了OPC分化和髓鞘形成的缺陷。同时,也观察到了表达ALDOC-K14R突变体的OPC中的mTORC1(mammalian/mechanistic target of rapamycin complex 1)也在脱髓鞘疾病模型中的OPC中被显著抑制,这解释了ALDOC的乙酰化对于维持mTORC1的活力,并进一步维持OPC增殖、分化以及髓鞘形成所必需的合成代谢通路的重要意义(图1)。他们还发现,只要溶酶体途径一直保持在 钝化 的状态,其它途径的AMPK的活化都不能有效地抑制mTORC1;也就是说,只有在溶酶体途径的AMPK激活才能有力地抑制mTORC1的活力,这也和先前在其它细胞和组织中所观察到的现象类似。
总之,通过 主动屏蔽 低糖信号,OPC确保了在葡萄糖缺乏的压力下,阻断本该被低糖诱导的AMPK的激活,从而确保了髓鞘修复这项 最高优先级 的任务仍能推进。这为我们未来开发旨在促进髓鞘再生的疗法,例如靶向ALDOC的乙酰化状态,提供了新的靶点和思路。
原文链接:
https://www.nature.com/articles/s42255-025-01386-8
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->